Using wrappper functions
Pascal Belleau, Astrid Deschênes and Alexander Krasnitz
Source:vignettes/Wrappers.Rmd
Wrappers.Rmd
Package: RAIDS
Authors: Pascal Belleau [cre, aut] (ORCID: https://orcid.org/0000-0002-0802-1071), Astrid Deschênes
[aut] (ORCID: https://orcid.org/0000-0001-7846-6749), David A. Tuveson
[aut] (ORCID: https://orcid.org/0000-0002-8017-2712), Alexander
Krasnitz [aut]
Version: 1.7.3
Compiled date: 2025-10-08
License: Apache License (>= 2)
This vignette explains, in further details, the used of the wrapper functions that were developed for a previous release of RAIDS.
While those functions are still working, we recommend using the new functions as described in the main vignette.
Main Steps
This is an overview of genetic ancestry inference from cancer-derived molecular data:
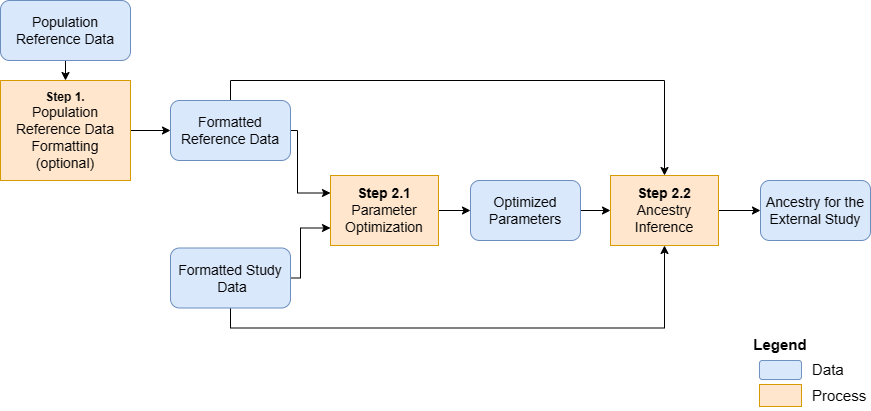
An overview of the genetic ancestry inference process.
The main steps are:
Step 1. Format reference data from the population reference dataset (optional)
Step 2.1 Optimize ancestry inference parameters
Step 2.2 Infer ancestry for the subjects of the external study
These steps are described in detail in the following. Steps 2.1 and 2.2 can be run together using one wrapper function.
Main Step - Ancestry Inference
A wrapper function encapsulates multiple steps of the workflow.
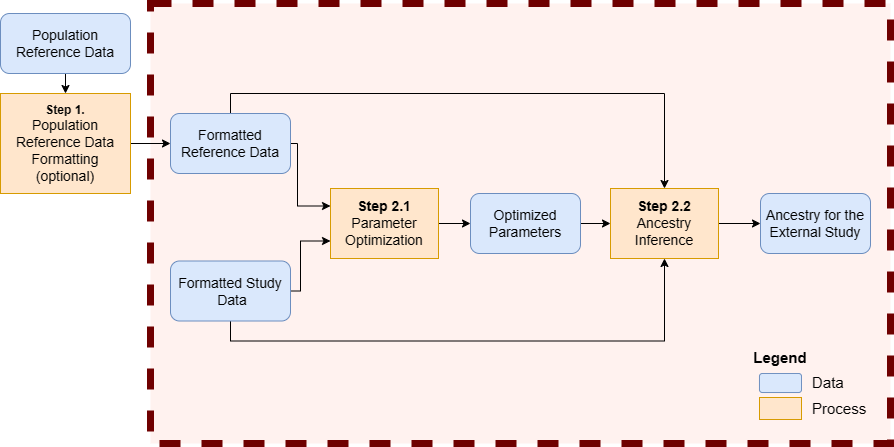
Final step - The wrapper function encapsulates multiple steps of the workflow.
In summary, the wrapper function generates the synthetic dataset and uses it to selected the optimal parameters before calling the genetic ancestry on the current profiles.
According to the type of input data (RNA or DNA), a specific wrapper function is available.
DNA Data - Wrapper function to run ancestry inference on DNA data
The wrapper function, called runExomeAncestry(), requires 4 files as input:
- The population reference GDS file
- The population reference SNV Annotation GDS file
- The Profile SNP file (one per sample present in the study)
- The Profile PED RDS file (one file with information for all profiles in the study)
In addition, a data.frame containing the general information about the study is also required. The data.frame must contain those 3 columns:
- study.id: The study identifier (example: TCGA-BRCA).
- study.desc: The description of the study.
- study.platform: The type of sequencing (example: RNA-seq).
Population reference files
For demonstration purpose, a small population reference GDS file (called ex1_good_small_1KG.gds) and a small population reference SNV Annotation GDS file (called ex1_good_small_1KG_Annot.gds) are included in this package. Beware that those two files should not be used to run a real ancestry inference.The results obtained with those files won’t be reliable.
The required population reference GDS file and population reference SNV Annotation GDS file should be stored in the same directory. In the example below, this directory is referred to as pathReference.
Profile SNP file
The Profile SNP file can be either in a VCF format or in a generic format.
The Profile SNP VCF file follows the VCF standard with at least those genotype fields: GT, AD and DP. The identifier of the genotype in the VCF file must correspond to the profile identifier Name.ID. The SNVs must be germline variants and should include the genotype of the wild-type homozygous at the selected positions in the reference. One file per profile is need and the VCF file must be gzipped.
Note that the name assigned to the Profile SNP VCF file has to correspond to the profile identifier Name.ID in the following analysis. For example, a SNP file called “Sample.01.vcf.gz” would be associated to the “Sample.01” profile.
A generic SNP file can replace the VCF file. The Profile SNP Generic file format is coma separated and the mandatory columns are:
- Chromosome: The name of the chromosome
- Position: The position on the chromosome
- Ref: The reference nucleotide
- Alt: The aternative nucleotide
- Count: The total count
- File1R: The count for the reference nucleotide
- File1A: The count for the alternative nucleotide
Beware that the starting position in the population reference GDS File is zero (like BED files). The Profile SNP Generic file should also start at position zero.
Note that the name assigned to the Profile SNP Generic file has to correspond to the profile identifier Name.ID in the following analysis. For example, a SNP file called “Sample.01.generic.txt.gz” would be associated to the “Sample.01” profile.
Profile PED RDS file
The Profile PED RDS file must contain a data.frame describing all the profiles to be analyzed. These 5 mandatory columns:
- Name.ID: The unique sample identifier. The associated profile SNP file should be called “Name.ID.txt.gz”.
- Case.ID: The patient identifier associated to the sample.
- Sample.Type: The information about the profile tissue source (primary tumor, metastatic tumor, normal, etc..).
- Diagnosis: The donor’s diagnosis.
- Source: The source of the profile sequence data (example: dbGAP_XYZ).
Important: The row names of the data.frame must be the profiles Name.ID.
This file is referred to as the Profile PED RDS file (PED for pedigree). Alternatively, the PED information can be saved in another type of file (CVS, etc..) as long as the data.frame information can be regenerated in R (with read.csv() or else).
Example
This example run an ancestry inference on an exome sample. Both population reference files are demonstration files and should not be used for a real ancestry inference. Beware that running an ancestry inference on real data will take longer to run.
#############################################################################
## Load required packages
#############################################################################
library(RAIDS)
library(gdsfmt)
## Path to the demo 1KG GDS file is located in this package
dataDir <- system.file("extdata", package="RAIDS")
#############################################################################
## Load the information about the profile
#############################################################################
data(demoPedigreeEx1)
head(demoPedigreeEx1)## Name.ID Case.ID Sample.Type Diagnosis Source
## ex1 ex1 ex1 Primary Tumor Cancer Databank B
#############################################################################
## The population reference GDS file and SNV Annotation GDS file
## need to be located in the same directory.
## Note that the population reference GDS file used for this example is a
## simplified version and CANNOT be used for any real analysis
#############################################################################
pathReference <- file.path(dataDir, "tests")
fileGDS <- file.path(pathReference, "ex1_good_small_1KG.gds")
fileAnnotGDS <- file.path(pathReference, "ex1_good_small_1KG_Annot.gds")
#############################################################################
## A data frame containing general information about the study
## is also required. The data frame must have
## those 3 columns: "study.id", "study.desc", "study.platform"
#############################################################################
studyDF <- data.frame(study.id="MYDATA",
study.desc="Description",
study.platform="PLATFORM",
stringsAsFactors=FALSE)
#############################################################################
## The Sample SNP VCF files (one per sample) need
## to be all located in the same directory.
#############################################################################
pathGeno <- file.path(dataDir, "example", "snpPileup")
#############################################################################
## Fix RNG seed to ensure reproducible results
#############################################################################
set.seed(3043)
#############################################################################
## Select the profiles from the population reference GDS file for
## the synthetic data.
## Here we select 2 profiles from the simplified 1KG GDS for each
## subcontinental-level.
## Normally, we use 30 profile for each
## subcontinental-level but it is too big for the example.
## The 1KG files in this example only have 6 profiles for each
## subcontinental-level (for demo purpose only).
#############################################################################
gds1KG <- snpgdsOpen(fileGDS)
dataRef <- select1KGPop(gds1KG, nbProfiles=2L)
closefn.gds(gds1KG)
## Seqinfo and BSgenome are required libraries to run this example
if (requireNamespace("Seqinfo", quietly=TRUE) &&
requireNamespace("BSgenome.Hsapiens.UCSC.hg38", quietly=TRUE)) {
## Chromosome length information
## chr23 is chrX, chr24 is chrY and chrM is 25
chrInfo <- Seqinfo::seqlengths(BSgenome.Hsapiens.UCSC.hg38::Hsapiens)[1:25]
###########################################################################
## The path where the Sample GDS files (one per sample)
## will be created needs to be specified.
###########################################################################
pathProfileGDS <- file.path(tempdir(), "exampleDNA", "out.tmp")
###########################################################################
## The path where the result files will be created needs to
## be specified
###########################################################################
pathOut <- file.path(tempdir(), "exampleDNA", "res.out")
## Example can only be run if the current directory is in writing mode
if (!dir.exists(file.path(tempdir(), "exampleDNA"))) {
dir.create(file.path(tempdir(), "exampleDNA"))
dir.create(pathProfileGDS)
dir.create(pathOut)
#########################################################################
## The wrapper function generates the synthetic dataset and uses it
## to selected the optimal parameters before calling the genetic
## ancestry on the current profiles.
## All important information, for each step, are saved in
## multiple output files.
## The 'genoSource' parameter has 2 options depending on how the
## SNP files have been generated:
## SNP VCF files have been generated:
## "VCF" or "generic" (other software)
##
#########################################################################
runExomeAncestry(pedStudy=demoPedigreeEx1, studyDF=studyDF,
pathProfileGDS=pathProfileGDS,
pathGeno=pathGeno,
pathOut=pathOut,
fileReferenceGDS=fileGDS,
fileReferenceAnnotGDS=fileAnnotGDS,
chrInfo=chrInfo,
syntheticRefDF=dataRef,
genoSource="VCF")
list.files(pathOut)
list.files(file.path(pathOut, demoPedigreeEx1$Name.ID[1]))
#######################################################################
## The file containing the ancestry inference (SuperPop column) and
## optimal number of PCA component (D column)
## optimal number of neighbours (K column)
#######################################################################
resAncestry <- read.csv(file.path(pathOut,
paste0(demoPedigreeEx1$Name.ID[1], ".Ancestry.csv")))
print(resAncestry)
## Remove temporary files created for this demo
unlink(pathProfileGDS, recursive=TRUE, force=TRUE)
unlink(pathOut, recursive=TRUE, force=TRUE)
unlink(file.path(tempdir(), "exampleDNA"), recursive=TRUE, force=TRUE)
}
}## sample.id D K SuperPop
## 1 ex1 14 6 AFRThe runExomeAncestry() function generates 3 types of files in the pathOut directory.
- The ancestry inference CSV file (“.Ancestry.csv” file)
- The inference information RDS file (“.infoCall.rds” file)
- The parameter information RDS files from the synthetic inference (“KNN.synt.*.rds” files in a sub-directory)
In addition, a sub-directory (named using the profile ID) is also created.
The inferred ancestry is stored in the ancestry inference CSV file (“.Ancestry.csv” file) which also contains those columns:
- sample.id: The unique identifier of the sample
- D: The optimal PCA dimension value used to infer the ancestry
- k: The optimal number of neighbors value used to infer the ancestry
- SuperPop: The inferred ancestry
RNA data - Wrapper function to run ancestry inference on RNA data
The process is the same as for the DNA but use the wrapper function called runRNAAncestry(). Internally the data is process differently. It requires 4 files as input:
- The population reference GDS file
- The population reference SNV Annotation GDS file
- The Profile SNP file (one per sample present in the study)
- The Profile PED RDS file (one file with information for all profiles in the study)
A data.frame containing the general information about the study is also required. The data.frame must contain those 3 columns:
- study.id: The study identifier (example: TCGA-BRCA).
- study.desc: The description of the study.
- study.platform: The type of sequencing (example: RNA-seq).
Population reference files
For demonstration purpose, a small population reference GDS file (called ex1_good_small_1KG.gds) and a small population reference SNV Annotation GDS file (called ex1_good_small_1KG_Annot.gds) are included in this package. Beware that those two files should not be used to run a real ancestry inference.The results obtained with those files won’t be reliable.
The required population reference GDS file and population reference SNV Annotation GDS file should be stored in the same directory. In the example below, this directory is referred to as pathReference.
Profile SNP file
The Profile SNP file can be either in a VCF format or in a generic format.
The Profile SNP VCF file follows the VCF standard with at least those genotype fields: GT, AD and DP. The identifier of the genotype in the VCF file must correspond to the profile identifier Name.ID. The SNVs must be germline variants and should include the genotype of the wild-type homozygous at the selected positions in the reference. One file per profile is need and the VCF file must be gzipped.
Note that the name assigned to the Profile SNP VCF file has to correspond to the profile identifier Name.ID in the following analysis. For example, a SNP file called “Sample.01.vcf.gz” would be associated to the “Sample.01” profile.
A generic SNP file can replace the VCF file. The Profile SNP Generic file format is coma separated and the mandatory columns are:
- Chromosome: The name of the chromosome
- Position: The position on the chromosome
- Ref: The reference nucleotide
- Alt: The aternative nucleotide
- Count: The total count
- File1R: The count for the reference nucleotide
- File1A: The count for the alternative nucleotide
Beware that the starting position in the population reference GDS File is zero (like BED files). The Profile SNP Generic file should also start at position zero.
Note that the name assigned to the Profile SNP Generic file has to correspond to the profile identifier Name.ID in the following analysis. For example, a SNP file called “Sample.01.generic.txt.gz” would be associated to the “Sample.01” profile.
Profile PED RDS file
The Profile PED RDS file must contain a data.frame describing all the profiles to be analyzed. These 5 mandatory columns:
-
Name.ID: The unique sample identifier. The associated
profile SNP file
should be called “Name.ID.txt.gz”. - Case.ID: The patient identifier associated to the sample.
- Sample.Type: The information about the profile tissue source (primary tumor, metastatic tumor, normal, etc..).
- Diagnosis: The donor’s diagnosis.
- Source: The source of the profile sequence data (example: dbGAP_XYZ).
Important: The row names of the data.frame must be the profiles Name.ID.
This file is referred to as the Profile PED RDS file (PED for pedigree). Alternatively, the PED information can be saved in another type of file (CVS, etc..) as long as the data.frame information can be regenerated in R (with read.csv() or else).
Example
This example run an ancestry inference on an RNA sample. Both population reference files are demonstration files and should not be used for a real ancestry inference. Beware that running an ancestry inference on real data will take longer to run.
#############################################################################
## Load required packages
#############################################################################
library(RAIDS)
library(gdsfmt)
## Path to the demo 1KG GDS file is located in this package
dataDir <- system.file("extdata", package="RAIDS")
#############################################################################
## Load the information about the profile
#############################################################################
data(demoPedigreeEx1)
head(demoPedigreeEx1)## Name.ID Case.ID Sample.Type Diagnosis Source
## ex1 ex1 ex1 Primary Tumor Cancer Databank B
#############################################################################
## The population reference GDS file and SNV Annotation GDS file
## need to be located in the same directory.
## Note that the population reference GDS file used for this example is a
## simplified version and CANNOT be used for any real analysis
#############################################################################
pathReference <- file.path(dataDir, "tests")
fileGDS <- file.path(pathReference, "ex1_good_small_1KG.gds")
fileAnnotGDS <- file.path(pathReference, "ex1_good_small_1KG_Annot.gds")
#############################################################################
## A data frame containing general information about the study
## is also required. The data frame must have
## those 3 columns: "study.id", "study.desc", "study.platform"
#############################################################################
studyDF <- data.frame(study.id="MYDATA",
study.desc="Description",
study.platform="PLATFORM",
stringsAsFactors=FALSE)
#############################################################################
## The Sample SNP VCF files (one per sample) need
## to be all located in the same directory.
#############################################################################
pathGeno <- file.path(dataDir, "example", "snpPileupRNA")
#############################################################################
## Fix RNG seed to ensure reproducible results
#############################################################################
set.seed(3043)
#############################################################################
## Select the profiles from the population reference GDS file for
## the synthetic data.
## Here we select 2 profiles from the simplified 1KG GDS for each
## subcontinental-level.
## Normally, we use 30 profile for each
## subcontinental-level but it is too big for the example.
## The 1KG files in this example only have 6 profiles for each
## subcontinental-level (for demo purpose only).
#############################################################################
gds1KG <- snpgdsOpen(fileGDS)
dataRef <- select1KGPop(gds1KG, nbProfiles=2L)
closefn.gds(gds1KG)
## Seqinfo and BSgenome are required libraries to run this example
if (requireNamespace("Seqinfo", quietly=TRUE) &&
requireNamespace("BSgenome.Hsapiens.UCSC.hg38", quietly=TRUE)) {
## Chromosome length information
## chr23 is chrX, chr24 is chrY and chrM is 25
chrInfo <- Seqinfo::seqlengths(BSgenome.Hsapiens.UCSC.hg38::Hsapiens)[1:25]
#############################################################################
## The path where the Sample GDS files (one per sample)
## will be created needs to be specified.
#############################################################################
pathProfileGDS <- file.path(tempdir(), "exampleRNA", "outRNA.tmp")
#############################################################################
## The path where the result files will be created needs to
## be specified
#############################################################################
pathOut <- file.path(tempdir(), "exampleRNA", "resRNA.out")
## Example can only be run if the current directory is in writing mode
if (!dir.exists(file.path(tempdir(), "exampleRNA"))) {
dir.create(file.path(tempdir(), "exampleRNA"))
dir.create(pathProfileGDS)
dir.create(pathOut)
#########################################################################
## The wrapper function generates the synthetic dataset and uses it
## to selected the optimal parameters before calling the genetic
## ancestry on the current profiles.
## All important information, for each step, are saved in
## multiple output files.
## The 'genoSource' parameter has 2 options depending on how the
## SNP files have been generated:
## SNP VCF files have been generated:
## "VCF" or "generic" (other software)
#########################################################################
runRNAAncestry(pedStudy=demoPedigreeEx1, studyDF=studyDF,
pathProfileGDS=pathProfileGDS,
pathGeno=pathGeno,
pathOut=pathOut,
fileReferenceGDS=fileGDS,
fileReferenceAnnotGDS=fileAnnotGDS,
chrInfo=chrInfo,
syntheticRefDF=dataRef,
blockTypeID="GeneS.Ensembl.Hsapiens.v86",
genoSource="VCF")
list.files(pathOut)
list.files(file.path(pathOut, demoPedigreeEx1$Name.ID[1]))
#########################################################################
## The file containing the ancestry inference (SuperPop column) and
## optimal number of PCA component (D column)
## optimal number of neighbours (K column)
#########################################################################
resAncestry <- read.csv(file.path(pathOut,
paste0(demoPedigreeEx1$Name.ID[1], ".Ancestry.csv")))
print(resAncestry)
## Remove temporary files created for this demo
unlink(pathProfileGDS, recursive=TRUE, force=TRUE)
unlink(pathOut, recursive=TRUE, force=TRUE)
unlink(file.path(tempdir(), "example"), recursive=TRUE, force=TRUE)
}
}## sample.id D K SuperPop
## 1 ex1 14 6 AFRThe runRNAAncestry() function generates 3 types of files in the pathOut directory.
- The ancestry inference CSV file (“.Ancestry.csv” file)
- The inference information RDS file (“.infoCall.rds” file)
- The parameter information RDS files from the synthetic inference (“KNN.synt.*.rds” files in a sub-directory)
In addition, a sub-directory (named using the profile ID) is also created.
The inferred ancestry is stored in the ancestry inference CSV file (“.Ancestry.csv” file) which also contains those columns:
- sample.id: The unique identifier of the sample
- D: The optimal PCA dimension value used to infer the ancestry
- k: The optimal number of neighbors value used to infer the ancestry
- SuperPop: The inferred ancestry
Format population reference dataset (optional)
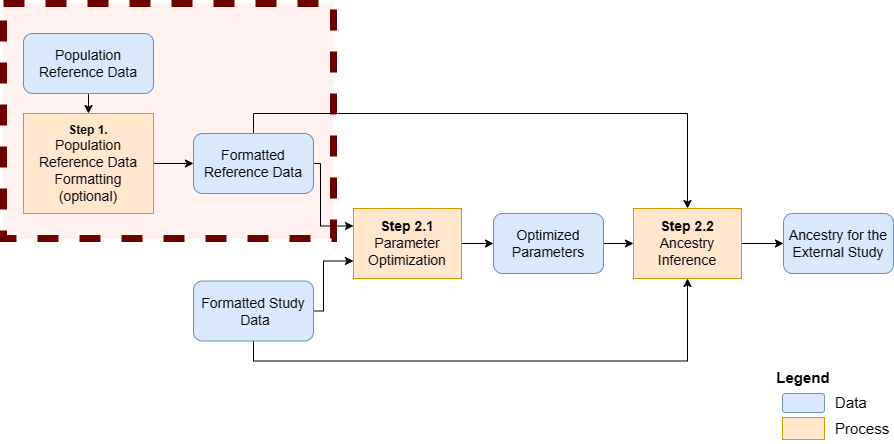
Step 1 - Formatting the information from the population reference dataset (optional)
A population reference dataset with known ancestry is required to infer ancestry.
Three important reference files, containing formatted information about the reference dataset, are required:
- The population reference GDS File
- The population reference SNV Annotation GDS file
- The population reference SNV Retained VCF file
The format of those files are described the Population reference dataset GDS files vignette.
The reference files associated to the Cancer Research associated paper are available. Note that these pre-processed files are for 1000 Genomes (1KG), in hg38. The files are available here:
https://labshare.cshl.edu/shares/krasnitzlab/aicsPaper
The size of the 1KG GDS file is 15GB.
The 1KG GDS file is mapped on hg38 (Lowy-Gallego et al. 2019).
This section can be skipped if you choose to use the pre-processed files.
Session info
Here is the output of sessionInfo() in the environment
in which this document was compiled:
## R version 4.5.1 (2025-06-13)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.3 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] Matrix_1.7-3 RAIDS_1.7.3 Rsamtools_2.25.3
## [4] Biostrings_2.77.2 XVector_0.49.1 GenomicRanges_1.61.5
## [7] IRanges_2.43.5 S4Vectors_0.47.4 Seqinfo_0.99.2
## [10] BiocGenerics_0.55.1 generics_0.1.4 dplyr_1.1.4
## [13] GENESIS_2.39.3 SNPRelate_1.43.1 gdsfmt_1.45.1
## [16] knitr_1.50 BiocStyle_2.37.1
##
## loaded via a namespace (and not attached):
## [1] RColorBrewer_1.1-3 jsonlite_2.0.0
## [3] shape_1.4.6.1 magrittr_2.0.4
## [5] jomo_2.7-6 GenomicFeatures_1.61.6
## [7] farver_2.1.2 logistf_1.26.1
## [9] nloptr_2.2.1 rmarkdown_2.30
## [11] fs_1.6.6 BiocIO_1.19.0
## [13] ragg_1.5.0 vctrs_0.6.5
## [15] memoise_2.0.1 minqa_1.2.8
## [17] RCurl_1.98-1.17 quantsmooth_1.75.0
## [19] htmltools_0.5.8.1 S4Arrays_1.9.1
## [21] curl_7.0.0 broom_1.0.10
## [23] pROC_1.19.0.1 SparseArray_1.9.1
## [25] mitml_0.4-5 sass_0.4.10
## [27] bslib_0.9.0 desc_1.4.3
## [29] sandwich_3.1-1 zoo_1.8-14
## [31] cachem_1.1.0 GenomicAlignments_1.45.4
## [33] lifecycle_1.0.4 iterators_1.0.14
## [35] pkgconfig_2.0.3 R6_2.6.1
## [37] fastmap_1.2.0 rbibutils_2.3
## [39] MatrixGenerics_1.21.0 digest_0.6.37
## [41] GWASTools_1.55.0 AnnotationDbi_1.71.1
## [43] textshaping_1.0.3 RSQLite_2.4.3
## [45] httr_1.4.7 abind_1.4-8
## [47] mgcv_1.9-3 compiler_4.5.1
## [49] bit64_4.6.0-1 S7_0.2.0
## [51] backports_1.5.0 BiocParallel_1.43.4
## [53] DBI_1.2.3 pan_1.9
## [55] MASS_7.3-65 quantreg_6.1
## [57] DelayedArray_0.35.3 rjson_0.2.23
## [59] DNAcopy_1.83.0 tools_4.5.1
## [61] lmtest_0.9-40 nnet_7.3-20
## [63] glue_1.8.0 restfulr_0.0.16
## [65] nlme_3.1-168 grid_4.5.1
## [67] gtable_0.3.6 operator.tools_1.6.3
## [69] BSgenome_1.77.2 formula.tools_1.7.1
## [71] class_7.3-23 tidyr_1.3.1
## [73] ensembldb_2.33.2 data.table_1.17.8
## [75] GWASExactHW_1.2 stringr_1.5.2
## [77] foreach_1.5.2 pillar_1.11.1
## [79] splines_4.5.1 lattice_0.22-7
## [81] survival_3.8-3 rtracklayer_1.69.1
## [83] bit_4.6.0 SparseM_1.84-2
## [85] BSgenome.Hsapiens.UCSC.hg38_1.4.5 tidyselect_1.2.1
## [87] SeqVarTools_1.47.0 reformulas_0.4.1
## [89] bookdown_0.45 ProtGenerics_1.41.0
## [91] SummarizedExperiment_1.39.2 RhpcBLASctl_0.23-42
## [93] xfun_0.53 Biobase_2.69.1
## [95] matrixStats_1.5.0 stringi_1.8.7
## [97] UCSC.utils_1.5.0 lazyeval_0.2.2
## [99] yaml_2.3.10 boot_1.3-31
## [101] evaluate_1.0.5 codetools_0.2-20
## [103] tibble_3.3.0 BiocManager_1.30.26
## [105] cli_3.6.5 rpart_4.1.24
## [107] systemfonts_1.3.1 Rdpack_2.6.4
## [109] jquerylib_0.1.4 Rcpp_1.1.0
## [111] GenomeInfoDb_1.45.12 png_0.1-8
## [113] XML_3.99-0.19 parallel_4.5.1
## [115] MatrixModels_0.5-4 ggplot2_4.0.0
## [117] pkgdown_2.1.3 blob_1.2.4
## [119] AnnotationFilter_1.33.0 bitops_1.0-9
## [121] lme4_1.1-37 glmnet_4.1-10
## [123] SeqArray_1.49.8 VariantAnnotation_1.55.1
## [125] scales_1.4.0 purrr_1.1.0
## [127] crayon_1.5.3 rlang_1.1.6
## [129] KEGGREST_1.49.1 mice_3.18.0