Using functional enrichment results in gprofiler2 format to create an enrichment map in an igraph format
Source:R/methodsEmap.R
createEnrichMapAsIgraph.RdUser selected enrichment terms are used to create an enrichment
map. The selection of the term can by specifying by the
source of the terms (GO:MF, REAC, TF, etc.) or by listing the selected
term IDs. The map is only generated when there is at least on
significant term to graph. The output is an igraph.
Usage
createEnrichMapAsIgraph(
gostObject,
query,
source = c("TERM_ID", "GO:MF", "GO:CC", "GO:BP", "KEGG", "REAC", "TF", "MIRNA", "HPA",
"CORUM", "HP", "WP"),
termIDs = NULL,
removeRoot = TRUE,
showCategory = 30L,
similarityCutOff = 0.2
)Arguments
- gostObject
a
listcorresponding to gprofiler2 enrichment output that contains and that contains the results from an enrichment analysis.- query
a
characterstring representing the name of the query that is going to be used to generate the graph. The query must exist in thegostObjectobject.- source
a
characterstring representing the selected source that will be used to generate the network. To hand-pick the terms to be used, "TERM_ID" should be used and the list of selected term IDs should be passed through thetermIDsparameter. The possible sources are "GO:BP" for Gene Ontology Biological Process, "GO:CC" for Gene Ontology Cellular Component, "GO:MF" for Gene Ontology Molecular Function, "KEGG" for Kegg, "REAC" for Reactome, "TF" for TRANSFAC, "MIRNA" for miRTarBase, "CORUM" for CORUM database, "HP" for Human phenotype ontology and "WP" for WikiPathways. Default: "TERM_ID".- termIDs
a
vectorofcharacterstrings that contains the term IDS retained for the creation of the network. Default:NULL.- removeRoot
a
logicalthat specified if the root terms of the selected source should be removed (when present). Default:TRUE.- showCategory
a positive
integerrepresenting the maximum number of terms to display. If ainteger, the firstnterms will be displayed. IfNULL, all terms will be displayed. Default:30L.- similarityCutOff
a positive
numericbetween 0 and 1 indicating the minimum level of similarity between two terms to have an edge linking the terms. Default:0.20.
Value
a igraph object which is the enrichment map for enrichment
results. The node have 2 attributes: "name" and "size". The "name"
corresponds to the term description. While the "size" corresponds to the
number of genes found in the specific gene set. The edges have
3 attributes: "similarity", "width", and "weight". All those 3 attributes
correspond to the Jaccard coefficient.
Examples
## Loading dataset containing result from an enrichment analysis done with
## gprofiler2
data(parentalNapaVsDMSOEnrichment)
## Extract query information (only one in this dataset)
query <- unique(parentalNapaVsDMSOEnrichment$result$query)
## Create graph for Gene Ontology - Cellular Component related results
mapG <- createEnrichMapAsIgraph(gostObject=parentalNapaVsDMSOEnrichment,
query=query, source="GO:CC", removeRoot=TRUE,
showCategory=30L, similarityCutOff=0.20)
## Required library igraph to show the graph
if(requireNamespace("igraph", quietly=TRUE)) {
## Using library igraph to show the graph
library(igraph)
plot(mapG)
}
#>
#> Attaching package: ‘igraph’
#> The following objects are masked from ‘package:stats’:
#>
#> decompose, spectrum
#> The following object is masked from ‘package:base’:
#>
#> union
 if (requireNamespace("ggplot2", quietly=TRUE) &&
requireNamespace("igraph", quietly=TRUE) &&
requireNamespace("ggtangle", quietly=TRUE) &&
requireNamespace("ggnetwork", quietly=TRUE)) {
## Using more complex set of libraries to display personalized graph
library(ggplot2)
library(igraph)
library(ggnetwork)
library(ggtangle)
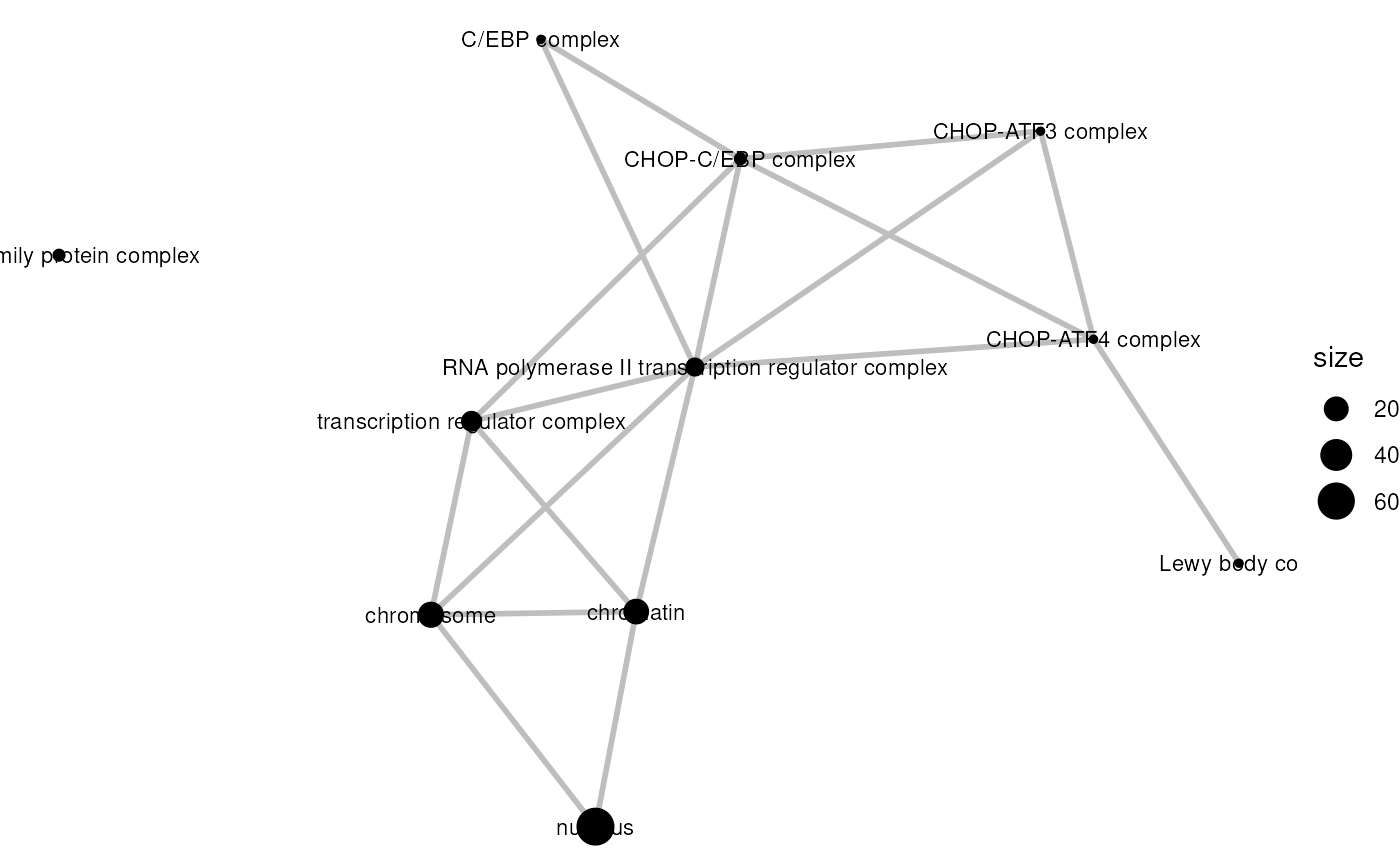
emapG <- ggplot(mapG, layout=layout_with_fr) +
geom_edge(color="gray", linewidth=1) +
geom_nodes(aes(size=size)) +
geom_nodetext(aes(label=name), color="black", size=3) +
theme_void()
emapG
}
#> Registered S3 method overwritten by 'ggnetwork':
#> method from
#> fortify.igraph ggtree
#> ggtangle v0.1.2 Learn more at https://yulab-smu.top/
#>
if (requireNamespace("ggplot2", quietly=TRUE) &&
requireNamespace("igraph", quietly=TRUE) &&
requireNamespace("ggtangle", quietly=TRUE) &&
requireNamespace("ggnetwork", quietly=TRUE)) {
## Using more complex set of libraries to display personalized graph
library(ggplot2)
library(igraph)
library(ggnetwork)
library(ggtangle)
emapG <- ggplot(mapG, layout=layout_with_fr) +
geom_edge(color="gray", linewidth=1) +
geom_nodes(aes(size=size)) +
geom_nodetext(aes(label=name), color="black", size=3) +
theme_void()
emapG
}
#> Registered S3 method overwritten by 'ggnetwork':
#> method from
#> fortify.igraph ggtree
#> ggtangle v0.1.2 Learn more at https://yulab-smu.top/
#>