Contents
- 1 License
- 2 Citing
- 3 Introduction
- 4 Installing and loading CNprep package
- 5 Processing copy number
-
6 Create and apply a mask to copy number events
- 6.1 Remove non-somatic copy number events when matching normal profile is available
- 6.2 Remove non-somatic copy number events when matching normal profile is not available but multiple cancer-free profiles are available
- 6.3 Identify recurrent copy number events in a cohort
- 6.4 Apply mask to a table of copy number events
- 7 Using parallelization
- 8 Reproducible research
- 9 Session info
- References
Package: CNprep
Authors: Alex Krasnitz [aut],
Pascal Belleau [aut, cre] (https://orcid.org/0000-0002-0802-1071),
Astrid Deschênes [aut] (https://orcid.org/0000-0001-7846-6749),
Guoli Sun [aut]
Version: 2.1.1
Compiled date: 2021-06-29
License: GPL-2
1 License
The CNprep package and the underlying CNprep code are distributed under the GPL-2 License. You are free to use and redistribute this software.
2 Citing
If you use this package for a publication, we ask you to cite the following:
Belleau P, Deschênes A, Sun G et al. CNprep: Copy number event detection [version 1; not peer reviewed]. F1000Research 2020, 9:743 (poster) (https://doi.org/10.7490/f1000research.1118065.1)
3 Introduction
The CNprep package has been implemented to identify, through a multi-step process, segments with significant copy number deviation from normal state. To this end, CNprep uses both DNA copy number initial form (copy number as a noisy function of genomic position, also known as bins) and their approximation by a piecewise-constant function (segmentation).
Copy number variations (CNVs) are associated with a wide spectrum of pathological conditions and complex traits, such as Tourette Disorder (Wang et al. 2018) and especially cancer (Pelttari et al. 2018; Franch-Expósito et al. 2018). Furthermore, CNVs can potentially be used as biomarkers for cancer diagnosis (Pan et al. 2018).
Since late 1990s, one commonly used technology to detect CNVs is the Comparative Genomic Hybridization (CGH) microarray (Carter 2007). From those microarrays, software such as DNACopy (Seshan and Olshen 2019) can identify regions of gained and lost copy number. However, because of the low resolution of the array, detection of short CNVs is limited (Duan et al. 2013). The availability and low cost of Next-Generation Sequencing (NGS) have enabled precise detection of CNVs for the entire genome with the exclusion of few hard to map regions. This lead to the development of software adapted to whole-genome and whole-exome sequencing, such as CNVkit (Talevich et al. 2016) and exomecopy (Love 2019).
In the analysis of bulk tumor DNA cells, not all cells share the same ploidy or harbor the same copy number alterations. Hence, segments identified may not share integer copy number. So, evaluating the value of the copy number corresponding to the normal state of the cells, relative to which somatic alterations occur, is difficult (Kendall and Krasnitz 2014).
The CNprep package has been implemented to identify,
TODO
4 Installing and loading CNprep package
The CNprep package can be installed from github website using devtools package:
library(devtools)
devtools::install_github("belleau/CNprep")As with any R package, the CNprep package has to be loaded with the following command:
6 Create and apply a mask to copy number events
In the CNprep package, the makeCNPmask() and applyCNPmask() functions are complementary methods.
The makeCNPmask() enables the identification of genomic regions of interest. Those regions can be of interest because those are regions are not specific to the analyzed condition(s), as an example, common copy number alterations that are not related to cancer should be removed when alterations happening in cancer tissues is of interest. Those regions can also be of interest because of their high recurrence in a specific cohort.
The applyCNPmask() function removes the regions identified by the makeCNPmask() function from the dataset.
6.1 Remove non-somatic copy number events when matching normal profile is available
Copy number alterations are not always specific to tumor tissues. Some alterations can also be present in the cancer-free tissues, those are called inherited alterations. Yet, somatic copy number alterations, specific to cancer, are often the events of interest.
In the CNprep package, the makeCNPmask() function has been implemented to identify all inherited alterations. This function requires the copy number profile of a matching normal tissue.
TODO explain how to
6.2 Remove non-somatic copy number events when matching normal profile is not available but multiple cancer-free profiles are available
When matching cancer-free profile is not available, the identification of inherited alterations is based on the population-wide inherited copy number data. To be able to do so, a cohort of cancer-free profiles is needed.
TODO explain how to
6.3 Identify recurrent copy number events in a cohort
When multiple bulk cancer tissues DNA profiles are available, the makeCNPmask() function can be used to identify the recurrent copy number.
The makeCNPmask() function takes as inputs:
- a table of copy number events with genomic positions for all the profiles
- an upper threshold
- a lower threshold
As a first step, interval regions where the event count is above the lower threshold are identified. Among those retained regions, the function only returns the interval regions that contain at least one sub-interval with the count above the upper threshold.
Hence, the final mask is a set of genomic intervals that passed both thresholds.
As an example, a mask in created from copy number events detected in 10 cancer samples. In Figure 1, the top section represent all copy number events detected in the 10 samples for a small region of chromosome 1. The middle section shows the frequency of the events with the lower and upper thresholds selected for this analysis. The bottom section displays the result of the makeCNPmask() function for this dataset when the lower threshold is set to 0.25 and upper threshold to 0.45.
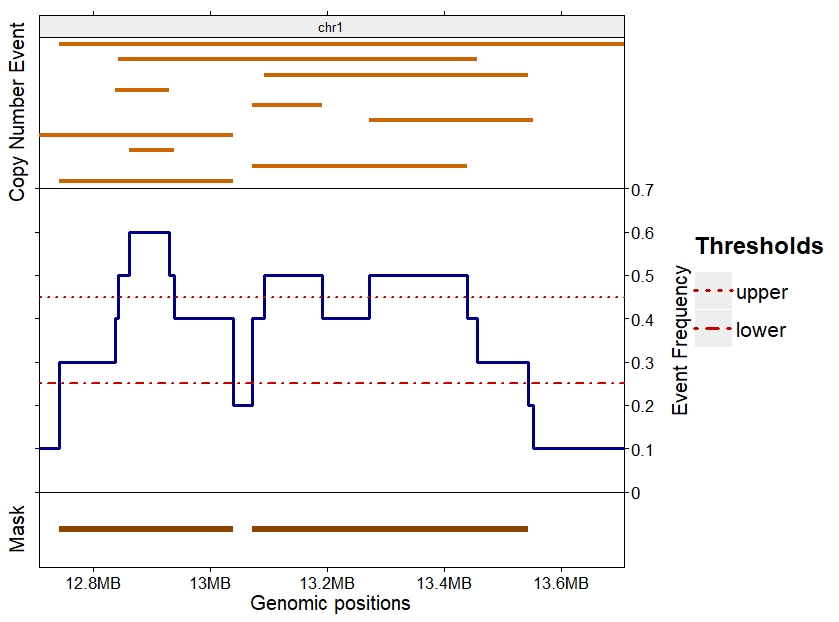
Figure 1: Example of makeCNPmask() function
Copy number events detected in 10 samples in top section. Frequency of the events with the selected lower and upper thresholds in the middle section. Result of the makeCNPmask() function for those thresholds in the bottom section.
Using an upper threshold of 0.55 on the same copy number events results in only one genomic region in the mask, as shown in Figure 2. This region is the only one to respect both lower and upper thresholds.
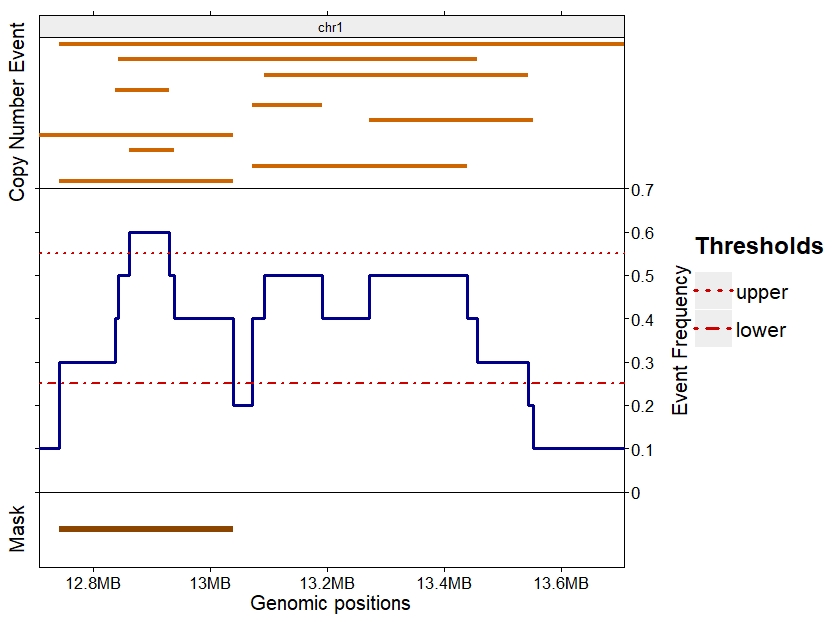
Figure 2: Example of makeCNPmask() function with stringent parameters
Copy number events detected in 10 samples in top section. Frequency of the events with the selected lower and upper thresholds in the middle section. Result of the makeCNPmask() function for those thresholds in the bottom section.
The following section demonstrates the usage of makeCNPmask() on a more complex dataset composed of 1203 breast cancer samples.
## Needed libraries
library(GenomicRanges)
## Loading required package: IRanges
## Loading required package: GenomeInfoDb
library(gtrellis)
## Loading required package: grid
## ========================================
## gtrellis version 1.24.0
## Bioconductor page: http://bioconductor.org/packages/gtrellis/
## Github page: https://github.com/jokergoo/gtrellis
## Documentation: http://bioconductor.org/packages/gtrellis/
##
## If you use it in published research, please cite:
## Gu, Z. gtrellis: an R/Bioconductor package for making genome-level
## Trellis graphics. BMC Bioinformatics 2016.
##
## This message can be suppressed by:
## suppressPackageStartupMessages(library(gtrellis))
## ========================================
## Load example containing the copy number events for 1203 samples
data(cnpexample)
## Retaining only the amplification events
ampCNP <- subset(cnpexample, copy.num == "amp")
## The amplification table has 4 columns:
## copy.num identifies the type of event
## chrom is the number identifying the chromosome
## chrom.start is the starting position of the event
## chrom.end is the ending position of the event
head(ampCNP, n=3)
## copy.num chrom chrom.start chrom.end
## 2 amp 1 12842827 13439275
## 5 amp 2 86198238 86575258
## 16 amp 5 772772 860239
## Create a mask using the amplification events.
## The name of the columns for the chromosome identification
## (chromCol parameter), the starting and ending positions
## (startCol and endCol parameters) must be given.
## The number of samples (nProf parameter) is also needed.
ampCNPmask <- makeCNPmask(imat=ampCNP, chromCol="chrom",
startCol="chrom.start", endCol="chrom.end", nProf=1203,
uThresh=0.2, dThresh=0.01)
## Transfrom the mask segments into Genomic Ranges
grAmpCNP <- GRanges(seqnames = Rle(paste0("chr", ampCNP$chrom)),
ranges = IRanges(start = ampCNP$chrom.start, end = ampCNP$chrom.end + 1),
strand = Rle(strand(c("*")), nrow(ampCNP)))
## Create trellis graph
gtrellis_layout(species = "hg19", category=paste0("chr", c(1,7,9,15)),
n_track=3, nrow=2, track_height=unit.c(2*grobHeight(textGrob("chr1")),
unit(10, "null"), unit(2, "null")))
## Add track for chromosome names
add_track(panel_fun = function(gr) {
chr <- get_cell_meta_data("name")
grid.rect(gp=gpar(fill="#EEEEEE"))
grid.text(chr) })
## Add segments representing the copy number events
add_segments_track(grAmpCNP, value=runif(length(grAmpCNP)),
gp=gpar(col="blue", lwd=4))
## Add segments representing the masks
maskFinal <- as.data.frame(ampCNPmask)
maskFinal$chrom <- paste0("chr", as.character(maskFinal$chrom))
add_rect_track(maskFinal, h1=0, h2=1, gp=gpar(col="red2", fill="red2"))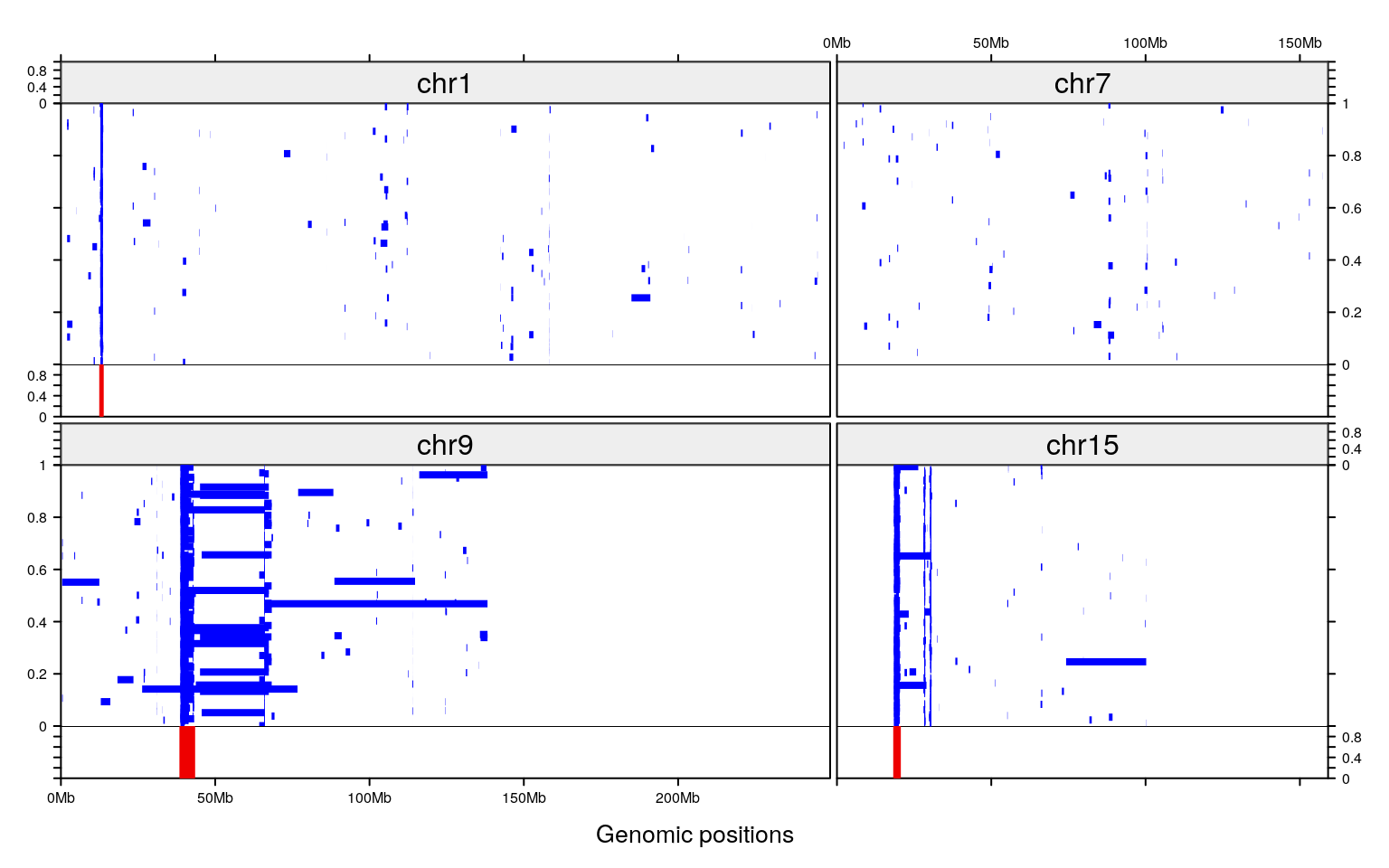
Figure 3: Example of makeCNPmask() on a dataset of 1203 samples
The amplification events are shown, in blue, in the top section. Results of the makeCNPmask() are shown for four chromosomes, in red, in the bottom section. In this example, only chromosome 7 doesn’t have any mask.
7 Using parallelization
For experiments with many samples, one can take advantage of parallelized computation. Parallelizing CNpreprocessing() function can easily be accomplished by setting the nJobs parameter to a number higher than one. Beware that the parallelization is done per sample. So, the nJobs value should not be higher than the number of samples in the dataset.
## Load needed datasets
data(segexample)
data(ratexample)
data(normsegs)
## The number of threads is set to 4 (nJobs parameter)
## Only the chromosomes 1 to 6 are analyzed (chromRange parameter)
segtable <- CNpreprocessing(segall=segexample, ratall=ratexample,
idCol="ID", startCol="start", endCol="end", chromCol="chrom",
bpStartCol="chrom.pos.start", bpEndCol="chrom.pos.end", blsize=50,
minJoin=0.25, cWeight=0.4, bsTimes=50, chromRange=1:6, nJobs=4,
normalLength=normsegs[,1], normalMedian=normsegs[,2])8 Reproducible research
To ensure reproducible results, set.seed() function should be call before CNpreprocessing(). Beware that the nJobs parameter must also be fixed. Change in the value of the nJobs parameter will lead to different results.
9 Session info
Here is the output of sessionInfo() on the system on which this document was compiled:
## R version 4.1.0 (2021-05-18)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 20.04.2 LTS
##
## Matrix products: default
## BLAS/LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.8.so
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=C
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] grid parallel stats4 stats graphics grDevices utils
## [8] datasets methods base
##
## other attached packages:
## [1] gtrellis_1.24.0 GenomicRanges_1.44.0 GenomeInfoDb_1.28.0
## [4] IRanges_2.26.0 CNprep_2.1.1 mclust_5.4.7
## [7] S4Vectors_0.30.0 BiocGenerics_0.38.0 BiocParallel_1.26.0
## [10] BiocStyle_2.20.2
##
## loaded via a namespace (and not attached):
## [1] compiler_4.1.0 BiocManager_1.30.16 highr_0.9
## [4] XVector_0.32.0 bitops_1.0-7 tools_4.1.0
## [7] zlibbioc_1.38.0 digest_0.6.27 evaluate_0.14
## [10] rlang_0.4.11 yaml_2.2.1 xfun_0.24
## [13] GenomeInfoDbData_1.2.6 stringr_1.4.0 knitr_1.33
## [16] GlobalOptions_0.1.2 GetoptLong_1.0.5 rmarkdown_2.9
## [19] bookdown_0.22 magrittr_2.0.1 htmltools_0.5.1.1
## [22] shape_1.4.6 circlize_0.4.13 colorspace_2.0-2
## [25] stringi_1.6.2 RCurl_1.98-1.3 crayon_1.4.1
## [28] rjson_0.2.20